
You and Peptides Research · ~10 min read
How Cells Read the Telegram
Receptor-ligand binding, GPCR cascades, and the picomolar problem
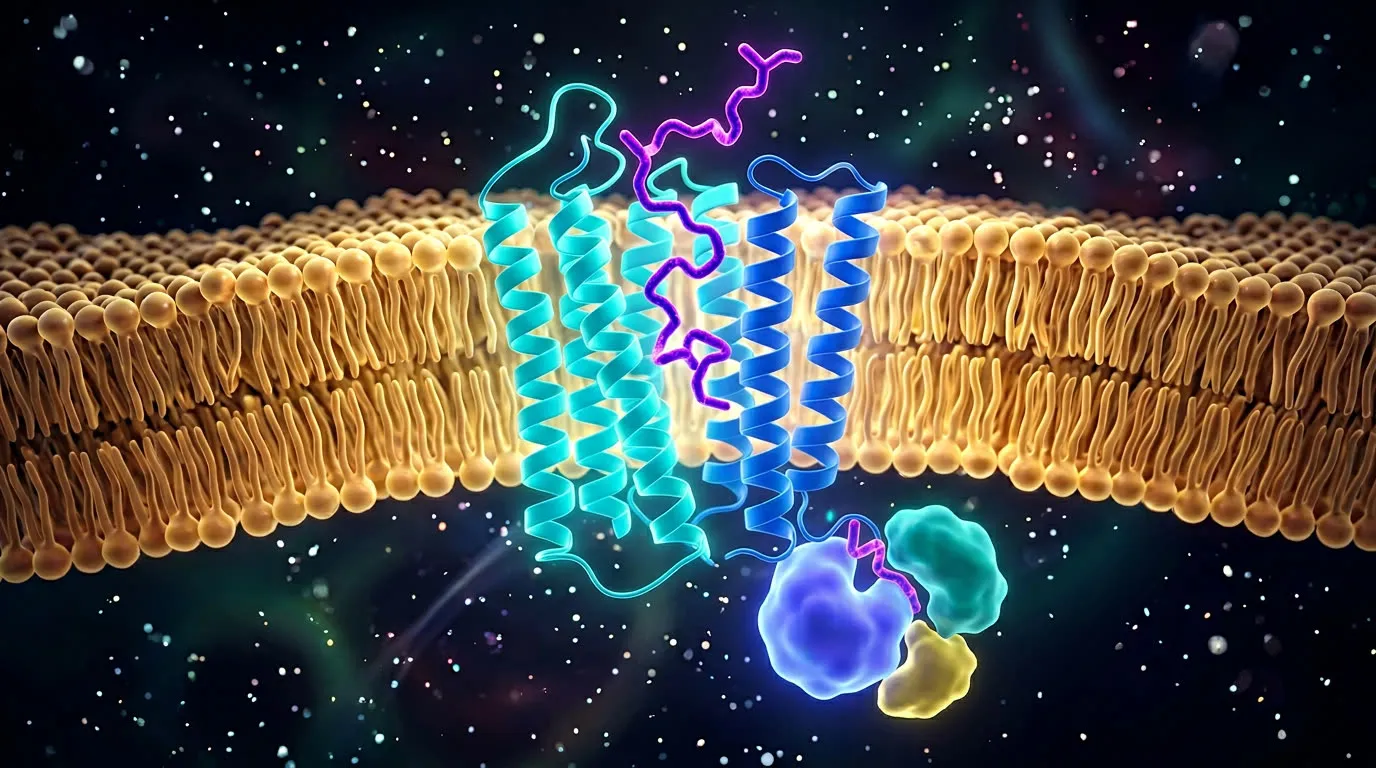
The big picture: specificity is everything
A typical human cell carries thousands of different receptors on its surface. Each is shaped to recognize a specific ligand — a hormone, neurotransmitter, growth factor, or signaling peptide — and to ignore everything else. This selectivity is the entire point. A drug that binds many receptors at once produces broad effects (and broad side effects). A drug that binds one receptor at high affinity produces precise effects.
Peptides are unusually good at this. Their three-dimensional shape is rich enough to fit a receptor pocket like a hand in a glove, but they are still small enough to manufacture and modify. The trade-off is fragility: peptides are easily destroyed by enzymes in the blood. Engineering around that fragility is what most modern peptide drug design is actually about.
GPCRs: the doorbell family
The single most important receptor family for peptide drugs is the G-protein coupled receptors (GPCRs). About a third of all approved drugs target a GPCR; the GLP-1 receptor, the GIP receptor, the ghrelin receptor, the somatostatin receptors, and dozens of others all belong to this family.
A GPCR is a single protein that snakes through the cell membrane seven times — its "seven transmembrane helices" — with a binding pocket facing outward and a tail facing inward. When a ligand binds the outside, the protein twists slightly, the tail changes shape, and an attached G-protein on the inside dissociates and triggers a cascade of downstream signals: cyclic AMP, calcium release, gene expression. One ligand binding event can amplify into thousands of downstream actions. It is the molecular doorbell.
Picomolar binding: what the numbers actually mean
Receptor binding affinity is measured by Kd — the concentration of ligand at which half the receptors are occupied. Smaller Kd means tighter binding. Modern GLP-1 agonists like semaglutide bind at picomolar concentrations: 10⁻¹² M, or one trillionth of a mole per liter. For comparison, a glass of seawater has roughly 10⁻⁵ M of dissolved sodium chloride, which is seven orders of magnitude higher.
Why does this matter? A drug that binds at picomolar concentrations only needs picomolar concentrations to work. That changes the entire dosing equation. Oral semaglutide has bioavailability of just 0.4–1%, but it does not need more than that. The molecule binds so tightly that even one one-hundredth of the original dose, surviving the gut and reaching the bloodstream, is therapeutically active.
The kon/koff balance: how long the doorbell rings
Affinity (Kd) is set by two rate constants: kon, how fast the ligand binds, and koff, how fast it dissociates. A drug with very high kon and very high koff has high affinity but only by spending a lot of time on and off the receptor — like a fast doorbell tap. A drug with moderate kon and very low koff has high affinity by staying bound for a long time once it lands — a long doorbell hold.
Most modern GLP-1 agonists are tuned for low koff. The ligand stays in the receptor for tens of minutes to hours after binding. This is why pharmacokinetic half-life and pharmacodynamic action don’t track perfectly: even after most of the drug has cleared from blood, the receptors that did get hit are still signaling.
Why DPP-4 destroys natural GLP-1 in 2 minutes
Native GLP-1 has a single weak spot at its N-terminus: the second residue, alanine, presents a peptide bond that fits perfectly into the active site of an enzyme called DPP-4 (dipeptidyl peptidase-4). DPP-4 cleaves the first two residues off the N-terminus, producing a stub that no longer binds the GLP-1 receptor.
The fix is sterically obvious in retrospect. Replace alanine with a residue that does not fit DPP-4’s active site, and the enzyme can no longer cleave. Semaglutide uses Aib (α-aminoisobutyric acid), which has two methyl groups at the α-carbon instead of one. DPP-4 simply cannot accept the substitution. Half-life jumps from 2 minutes to ~46 hours, and the rest of the molecule’s engineering (the fatty-acid albumin anchor) takes it from there to ~165 hours of receptor-bound time.
Designing for selectivity vs. designing for promiscuity
Sometimes the goal is to bind one receptor and only one. Selank, for instance, is engineered for specificity to certain neuropeptide pathways. Sometimes the goal is the opposite. Tirzepatide is a deliberately promiscuous molecule that binds both the GLP-1 receptor and the GIP receptor at therapeutic affinities. The dual activation produces additive effects on insulin secretion, weight loss, and gastric emptying — and is what made tirzepatide the first peptide therapy to break 20% body-weight loss in clinical trials.
The next generation of "balanced" agonists explicitly tunes the relative activity at multiple receptors. Retatrutide adds glucagon-receptor activity on top of GLP-1/GIP. The engineering goal is no longer "bind tightly to one thing" but "bind to N things in this exact ratio." That is a much harder design problem, and it is why AI structure-prediction tools are quickly becoming essential.